
| Table of contents |
Once PWTK is installed and configured (see Installation and Configuration Tutorial), we are ready to start using PWTK. It can be used interactively, but more often it is used to execute scripts. Among the first things to learn is how to specify QE input data and how to run QE programs in PWTK scripts.
There are two ways how to specify input data for QE executables in PWTK scripts.
Input data can be loaded into PWTK from existing QE input files using
the load_from
command. For example, an existing pw.x input file
(say pw.Si.in) can be loaded as:
load_from pw.x pw.Si.inHence, the usage of the load_from command is:
load_from program inputFile
where program is the name of the QE executable (e.g. pw.x, ph.x, pp.x) and inputFile is the name of the input file.
Alternatively, the pw.x input file can be loaded with the load_fromPWI command as:
load_fromPWI pw.Si.inwhere PWI stands for PW.x
Input. For other executables, analogous commands
exist (e.g. load_fromPHI, load_fromPPI,
load_fromNEBI). The
load_fromXXI (XXI = PWI, PHI,
PPI, NEBI…) commands exist for all explicitly supported QE programs in PWTK. Likewise, only the inputs for
the explicitly supported QE programs can be
loaded into PWTK with the
load_from command. To get the list of explicitly
supported programs, enter the PWTK
interactive prompt (by typing pwtk in the terminal) and
type report programs.
The second way of specifying input data is by entering them directly. The syntax is analogous to that of QE with the difference that namelists and cards are in encapsulated in curly braces (or double quotes), e.g.:
|
pw.x input file |
PWTK script |
|
|
BEWARE: PWTK
scripts are case-sensitive, implying that also
namelists, namelist variables, and cards are case-sensitive.
Namelists and cards usually have the same names as in QE but are written in
uppercase. Among exceptions are the generic
namelists (e.g. INPUT and INPUTPP) that are used by several QE programs, which are
renamed because PWTK requires
unique names. To list all exceptions, enter the PWTK interactive prompt (by typing
pwtk in the terminal) and type:
report exceptionswhich lists all the namelist and card naming exceptions in PWTK, i.e.:
-------------------------------------------------------------------------------
executable type QE-name PWTK-name
-------------------------------------------------------------------------------
cp.x supercard AUTOPILOT/ENDRULES AUTOPILOT
cppp.x namelist INPUTPP CPPP
d3hess.x namelist INPUT D3HESS
dynmat.x namelist INPUT DYNMAT
... ... ... ...For further info about input data specs in PWTK, see section 2.2 Specifying Input Data.
In PWTK, input data can be changed on the fly. For example, consider the following snippet:
SYSTEM {
nspin = 2
tot_magnetization = 1
}
SYSTEM {
nspin = 1
tot_magnetization = 0
} The current value of variables is now nspin = 1 and
tot_magnetization = 0. But for nspin=1, we
do not need to specify tot_magnetization. In PWTK, a defined namelist variable can
be deleted as follows:
SYSTEM {
tot_magnetization =
}In PWTK, a few namelist
variables are inherited from a “parent” namelist to “descendant”
namelists. Examples of such variables are the prefix
and outdir variables of the CONTROL namelist, INPUTPH’s
fildyn, and flfrc of the
q2r.x program. Hence, for example, if the
prefix and outdir variables are set in the
CONTROL namelist, their definition in the “descendant” namelists
(INPUTPP, INPUTPH, PROJWFC…) can be omitted because PWTK automatically sets them. Here is
an example:
CONTROL {
prefix = 'example1'
outdir = '.'
}
# 'outdir' and 'prefix' are inherited from the CONTROL namelist;
# no need to set them here
PROJWFC {
ngauss = 0
degauss = 0.01
DeltaE = 0.1
}An easy way to learn how to specify input data in PWTK is by using its
qe2pwtk program, which converts the QE input into the PWTK script. The usage is:
qe2pwtk ?-p program? inputFile
where the pair of question marks (?…?) indicates optional
arguments, program is the name of QE executable and
inputFile is the input file. For example, a
neb.x input file (say, neb.H2+H.in)
can be converted to PWTK syntax
as:
qe2pwtk -p neb.x neb.H2+H.inwhich gives:
PATH {
CI_scheme = 'auto'
num_of_images = 7
nstep_path = 100
string_method = 'neb'
}
CONTROL {
prefix = 'H2+H'
}
SYSTEM {
ecutwfc = 20.0
ibrav = 0
nat = 3
nspin = 2
ntyp = 1
tot_magnetization = 1.0
}
ELECTRONS {
conv_thr = 1.e-8
}
ATOMIC_SPECIES {
H 1.00794 H.pbe-rrkjus.UPF
}
CELL_PARAMETERS bohr {
12.000000000000000 0.000000000000000 0.000000000000000
0.000000000000000 5.000000000000000 0.000000000000000
0.000000000000000 0.000000000000000 5.000000000000000
}
K_POINTS gamma
POSITIONS {
FIRST_IMAGE
ATOMIC_POSITIONS bohr
H -5.274278671000 0.000000000000 0.000000000000 1 0 0
H 0.000000000000 0.000000000000 0.000000000000 0 0 0
H 1.490737029000 0.000000000000 0.000000000000 1 0 0
LAST_IMAGE
ATOMIC_POSITIONS bohr
H -1.490737127000 0.000000000000 0.000000000000
H 0.000000000000 0.000000000000 0.000000000000
H 5.274284748000 0.000000000000 0.000000000000
}Note that for neb.x, the PWTK syntax is slightly simpler than the QE syntax because its omits the BEGIN- and END-type keywords.
Analogously to specifying input data, there are two ways of how to run QE executables in PWTK.
They can be run by using the existing QE input files with the run command, i.e.:
run pw.x pw.Si.inwhich is equivalent to the following shell command:
$PREFIX pw.x < pw.Si.in > pw.Si.outwhere pw.Si.in is an existing pw.x
input file and the value of PREFIX is, e.g.,
mpirun -np 4. The run
command is similar to the Tcl exec
command but takes into account the PWTK specs, such as bin_dir,
prefix,
postfix…
The second way of running QE executables is by
first loading the input data into PWTK and then using the runXX
command. The advantage of loading the input data into PWTK is that one can change them on
the fly. For example:
tcl load_from pw.x pw.Si.in SYSTEM { ecutwfc = 25.0 } runXX pw.x pw.Si-e25.in
Alternatively, the pw.x program can be run with the
runPW
command as: runPW pw.Si-e25.in Analogous commands exist
for other QE
programs (e.g., runPH, runPP,
runNEB).
BEWARE: load_from
and runXX
commands are available only for explicitly supported QE programs in PWTK. To get the list of them, enter
the PWTK interactive prompt (by
typing pwtk in the terminal) and type:
report programswhich gives:
-------------------------------------------------------------------------------
executable runCmd rerunCmd input namespace
-------------------------------------------------------------------------------
all_currents.x runALL_CURRENTS ::pwtk::all_curri
band_interpolation.x runBAND_INTERPOLATION ::pwtk::band_inti
bands.x runBANDS ::pwtk::bi
bgw2pw.x runBGW2PW ::pwtk::bgw2pwi
cp.x runCP rerunCP ::pwtk::cpi
cppp.x runCPPP ::pwtk::cpppi
d3hess.x runD3HESS ::pwtk::d3hessi
dos.x runDOS ::pwtk::di
dynmat.x runDYNMAT ::pwtk::dmi
hp.x runHP ::pwtk::hpi
kcw.x runKCW ::pwtk::kcwi
ld1.x runLD1 ::pwtk::ld1i
matdyn.x runMATDYN ::pwtk::mdi
molecularpdos.x runMOPDOS ::pwtk::mpdi
neb.x runNEB rerunNEB ::pwtk::nebi
oscdft_et.x runOSCDFT_ET ::pwtk::oscdft_eti
oscdft_pp.x runOSCDFT_PP ::pwtk::oscdft_ppi
ph.x runPH rerunPH ::pwtk::phi
postahc.x runPOSTAHC ::pwtk::postahci
pp.x runPP ::pwtk::ppi
ppacf.x runPPACF ::pwtk::ppacfi
pprism.x runPPRISM ::pwtk::pprismi
projwfc.x runPROJWFC ::pwtk::pri
pw.x runPW rerunPW ::pwtk::pwi
pw2bgw.x runPW2BGW ::pwtk::pw2bgwi
pw2gw.x runPW2GW ::pwtk::pw2gwi
pw2wannier90.x runPW2WANNIER90 ::pwtk::pw2wan90i
pwcond.x runPWCOND rerunPWCOND ::pwtk::pwcondi
q2r.x runQ2R ::pwtk::q2ri
turbo_davidson.x runDAVIDSON rerunDAVIDSON ::pwtk::davi
turbo_eels.x runEELS rerunEELS ::pwtk::eelsi
turbo_lanczos.x runLANCZOS rerunLANCZOS ::pwtk::lani
turbo_magnon.x runMAGNON rerunMAGNON ::pwtk::magi
turbo_spectrum.x runSPECTRUM ::pwtk::speci
-------------------------------------------------------------------------------As seen above, loading input data into PWTK allows for changing them on the
fly, which is a handy feature that, among others, makes
parameter-scan calculations quite easy to script. For example, the
following script loads the pw.Si.in input
files and makes a series of pw.x calculations where
the value of the ecutwfc variable (i.e., kinetic energy
wavefunction cutoff) is scanned:
load_fromPWI pw.Si.in
scanpar ecut [seq 12 4 40] {
SYSTEM " ecutwfc = $ecut "
write ecutwfc.dat [pwo_totene [runPW pw.Si-e$ecut.in]]
}
plot -xr 10:40 -yf %.3f -xl "ecutwfc (Ry)" -yl "Total energy (Ry)" ecutwfc.datIn this script, the following new PWTK commands are used:
ecutwfc.dat datafile as
shown belowIn addition to performing the scan, this script also extracts the
total energy from the pw.x output file and writes
it to the ecutwfc.dat file, which is plotted at the end
with the plot
command. Let us save the above script into the
ecut.pwtk file and run it from the terminal as:
pwtk ecut.pwtkAfter the calculations are completed, we obtain the following plot:

Input-data stacking is a useful mechanism when performing multiple calculations, and we do not want input data modification for the preceding calculation to affect the input data for subsequent calculations. The concept is graphically presented here:
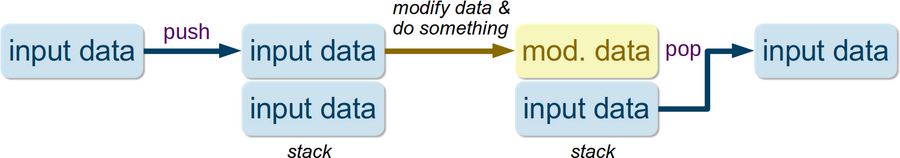
Here is the PWTK script example to illustrate the point; it utilizes the input_pushpop command which provides the stacking mechanism (read the comments):
load_fromPWI pw.Si.in
# the value of 'ecutwfc' in 'pw.Si.in' is 18.0 Ry;
# let's perform the calculation at higher 'ecutwfc'
input_pushpop {
SYSTEM { ecutrho = 30.0 }
# now the value of ecutrho = 30.0 Ry
runPW pw.Si-e30.0.in
}
# here, the value of ecutrho = 18.0 Ry again
# K_POINTS specs in 'pw.Si.in' are 4x4x4;
# let's perform the calculation with a denser K-grid
input_pushpop {
K_POINTS (automatic) {
8 8 8 1 1 1
}
runPW pw.Si-k8x8x8.in
}
# here K_POINTS specs are 4x4x4 again To further illustrate the input-data stacking mechanism, let us explain the scanpar command used above. It is similar to a one-parameter Tcl foreach loop but, in addition, uses the input-data stacking mechanism and, in combination with the write command, keeps track of the scanned parameter. Here is a comparison between equivalent script snippets using the scanpar and foreach commands:
| using scanpar | using foreach |
|
|
This implies that all input data modifications performed inside
the scanpar
loop are removed after the loop finishes. Note also that within the
scanpar
loop, the $ecut variable is not specified on the write
command line because write
knows which parameter(s) is (are) being scanned and adds them
automatically.
With the import
command it is possible to include a PWTK script into another script. This
command can be exploited for kind-of project-based configuration,
which helps systematizing its calculations. The idea is to create a
project common file (let us name it common.pwtk), where
default input data for the project are stored and then project
scripts import it.
Let us say that we want to study the adsorption of small organic
molecules (containing H, C, N, and O atoms) on low Miller index Rh
surfaces. After performing the basic convergence tests calculations
(as to determine the energy cutoff, lattice parameter, k-point
sampling…), we can create the project common.pwtk,
which could look something like this:
# default pw.x input data
CONTROL {
calculation = 'relax'
nstep = 400
}
SYSTEM {
ibrav = 0
ecutwfc = 50.
ecutrho = 400.
occupations = 'smearing'
degauss = 0.02
smearing = 'm-v'
}
ELECTRONS {
conv_thr = 1d-9
mixing_beta = 0.3
}
IONS {
upscale = 100.
}
ATOMIC_SPECIES {
H 1.0079 H.pbe-rrkjus_psl.1.0.0.UPF
C 12.011 C.pbe-n-rrkjus_psl.1.0.0.UPF
N 14.0067 N.pbe-n-rrkjus_psl.1.0.0.UPF
O 15.9994 O.pbe-n-rrkjus_psl.1.0.0.UPF
Rh 102.906 Rh.pbe-spn-rrkjus_psl.1.0.0.UPF
}
# default k-points for Rh(111)-2x2 & Rh(100)-2x2
K_POINTS (automatic) {
6 6 1 1 1 1
}
# default ph.x input data
INPUTPH {
tr2_ph = 1e-16
trans = .true.
nogg = .true.
alpha_mix(1) = 0.3
} {
0.0 0.0 0.0
}
# default projwfc.x input data
PROJWFC {
ngauss = 0
degauss = 0.01
DeltaE = 0.1
}REMARK: Note that all the pseudopotentials
used in the project are listed in the ATOMIC_SPECIES card, and the
SYSTEM’s ntyp variable is not assigned. For specific
cases, PWTK uses only
pseudopotentials for atomic types present in the ATOMIC_POSITIONS
card and sets the ntyp variable accordingly.
Then from specific scripts we can include this
common.pwtk file. For example, let us perform some
calculations in the Rh100-2x2-4L/ subdirectory. In
particular, we scan the lateral position of CH~3 over Rh(100)-2x2-4L
slab (4L = four-layer). The script could look something like:
import ../common.pwtk
# load the CH3 @ Rh(100)-2x2-4L structure (4L = 4-layers)
CELL_PARAMETERS_and_ATOMIC_POSITIONS_fromXSF CH3-Rh100-2x2-4L.xsf
# fix the bottom-layer Rh atoms (i.e. atom from 1 to 4)
fixAtoms 1-4
# fix the C atom (atom 17) laterally
fixAtoms 17 "0 0 1"
# the Rh-Rh distance is 2.687 Å
set dRhRh 2.687
# scan the CH3 laterally
scanpar dx [seq 0 0.2 1] {
scanpar dy [seq 0 0.2 1] {
set name CH3-$dx,$dy@Rh100-2x2-4L
outdir_postfix $name
CONTROL " prefix = '$name' "
# convert dx,dy to Å
set dx [expr $dx*$dRhRh]
set dy [expr $dy*$dRhRh]
# CH3 atoms are atoms from 17 to 20
displaceAtoms "$dx $dy 0.0" 17-20
write CH3.dat [pwo_totene [runPW relax.$name]]
}
}
# plot the result as a 3D surface-plot
splot -xl "Δx (Å)" -yl "Δy (Å)" -xf %.1f -yf %.1f -zf %.3f -t png CH3.datIn this script, we used the following new PWTK commands:
outdir as
outdir_prefix/outdir_postfixAfter the script finishes all calculations, splot
creates the following CH3.png image:
TODO–insert-splot
To learn more about PWTK scripting, please check its collection of online hyperlinked examples, which are documented with explanatory comments.
For further info, see also section 2. User Interface, which explains various aspects of PWTK. It is also worth checking the built-in workflows.